fix pimd command¶
Syntax¶
fix ID group-ID pimd keyword value ...
ID, group-ID are documented in fix command
pimd = style name of this fix command
zero or more keyword/value pairs may be appended
keyword = method or fmass or sp or temp or nhc
method value = pimd or nmpimd or cmd fmass value = scaling factor on mass sp value = scaling factor on Planck constant temp value = temperature (temperarate units) nhc value = Nc = number of chains in Nose-Hoover thermostat
Examples¶
fix 1 all pimd method nmpimd fmass 1.0 sp 2.0 temp 300.0 nhc 4
Description¶
This command performs quantum molecular dynamics simulations based on the Feynman path integral to include effects of tunneling and zero-point motion. In this formalism, the isomorphism of a quantum partition function for the original system to a classical partition function for a ring-polymer system is exploited, to efficiently sample configurations from the canonical ensemble (Feynman). The classical partition function and its components are given by the following equations:
The interested user is referred to any of the numerous references on this methodology, but briefly, each quantum particle in a path integral simulation is represented by a ring-polymer of P quasi-beads, labeled from 1 to P. During the simulation, each quasi-bead interacts with beads on the other ring-polymers with the same imaginary time index (the second term in the effective potential above). The quasi-beads also interact with the two neighboring quasi-beads through the spring potential in imaginary-time space (first term in effective potential). To sample the canonical ensemble, a Nose-Hoover massive chain thermostat is applied (Tuckerman). With the massive chain algorithm, a chain of NH thermostats is coupled to each degree of freedom for each quasi-bead. The keyword temp sets the target temperature for the system and the keyword nhc sets the number Nc of thermostats in each chain. For example, for a simulation of N particles with P beads in each ring-polymer, the total number of NH thermostats would be 3 x N x P x Nc.
Note
This fix implements a complete velocity-verlet integrator combined with NH massive chain thermostat, so no other time integration fix should be used.
The method keyword determines what style of PIMD is performed. A value of pimd is standard PIMD. A value of nmpimd is for normal-mode PIMD. A value of cmd is for centroid molecular dynamics (CMD). The difference between the styles is as follows.
In standard PIMD, the value used for a bead’s fictitious mass is arbitrary. A common choice is to use Mi = m/P, which results in the mass of the entire ring-polymer being equal to the real quantum particle. But it can be difficult to efficiently integrate the equations of motion for the stiff harmonic interactions in the ring polymers.
A useful way to resolve this issue is to integrate the equations of motion in a normal mode representation, using Normal Mode Path-Integral Molecular Dynamics (NMPIMD) (Cao1). In NMPIMD, the NH chains are attached to each normal mode of the ring-polymer and the fictitious mass of each mode is chosen as Mk = the eigenvalue of the Kth normal mode for k > 0. The k = 0 mode, referred to as the zero-frequency mode or centroid, corresponds to overall translation of the ring-polymer and is assigned the mass of the real particle.
Motion of the centroid can be effectively uncoupled from the other normal modes by scaling the fictitious masses to achieve a partial adiabatic separation. This is called a Centroid Molecular Dynamics (CMD) approximation (Cao2). The time-evolution (and resulting dynamics) of the quantum particles can be used to obtain centroid time correlation functions, which can be further used to obtain the true quantum correlation function for the original system. The CMD method also uses normal modes to evolve the system, except only the k > 0 modes are thermostatted, not the centroid degrees of freedom.
The keyword fmass sets a further scaling factor for the fictitious masses of beads, which can be used for the Partial Adiabatic CMD (Hone), or to be set as P, which results in the fictitious masses to be equal to the real particle masses.
The keyword sp is a scaling factor on Planck’s constant, which can be useful for debugging or other purposes. The default value of 1.0 is appropriate for most situations.
The PIMD algorithm in LAMMPS is implemented as a hyper-parallel scheme as described in (Calhoun). In LAMMPS this is done by using multi-replica feature in LAMMPS, where each quasi-particle system is stored and simulated on a separate partition of processors. The following diagram illustrates this approach. The original system with 2 ring polymers is shown in red. Since each ring has 4 quasi-beads (imaginary time slices), there are 4 replicas of the system, each running on one of the 4 partitions of processors. Each replica (shown in green) owns one quasi-bead in each ring.
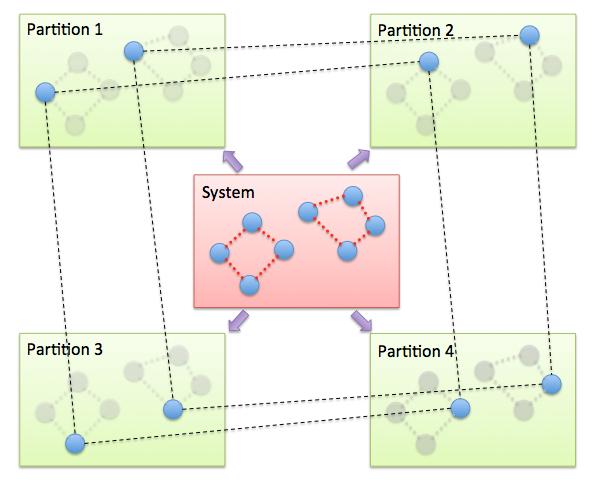
To run a PIMD simulation with M quasi-beads in each ring polymer using N MPI tasks for each partition’s domain-decomposition, you would use P = MxN processors (cores) and run the simulation as follows:
mpirun -np P lmp_mpi -partition MxN -in script
Note that in the LAMMPS input script for a multi-partition simulation, it is often very useful to define a uloop-style variable such as
variable ibead uloop M pad
where M is the number of quasi-beads (partitions) used in the calculation. The uloop variable can then be used to manage I/O related tasks for each of the partitions, e.g.
dump dcd all dcd 10 system_${ibead}.dcd
restart 1000 system_${ibead}.restart1 system_${ibead}.restart2
read_restart system_${ibead}.restart2
Restrictions¶
This fix is part of the REPLICA package. It is only enabled if LAMMPS was built with that package. See the Build package page for more info.
A PIMD simulation can be initialized with a single data file read via the read_data command. However, this means all quasi-beads in a ring polymer will have identical positions and velocities, resulting in identical trajectories for all quasi-beads. To avoid this, users can simply initialize velocities with different random number seeds assigned to each partition, as defined by the uloop variable, e.g.
velocity all create 300.0 1234${ibead} rot yes dist gaussian
Default¶
The keyword defaults are method = pimd, fmass = 1.0, sp = 1.0, temp = 300.0, and nhc = 2.
(Feynman) R. Feynman and A. Hibbs, Chapter 7, Quantum Mechanics and Path Integrals, McGraw-Hill, New York (1965).
(Tuckerman) M. Tuckerman and B. Berne, J Chem Phys, 99, 2796 (1993).
(Cao1) J. Cao and B. Berne, J Chem Phys, 99, 2902 (1993).
(Cao2) J. Cao and G. Voth, J Chem Phys, 100, 5093 (1994).
(Hone) T. Hone, P. Rossky, G. Voth, J Chem Phys, 124, 154103 (2006).
(Calhoun) A. Calhoun, M. Pavese, G. Voth, Chem Phys Letters, 262, 415 (1996).